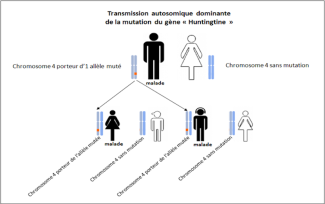
Huntington’s disease is a hereditary neurological condition caused by CAG expansion in the gene coding for the huntingtin protein. Its mode of transmission is autosomal dominant. In other words, inheriting the mutated copy of the Huntingtin gene (HTT gene) is enough to develop Huntington’s disease. The likelihood of a person with the disease passing on the expansion to their children is 50%, with a high risk of developing symptoms during their adult life.
Causes of Huntington’s disease
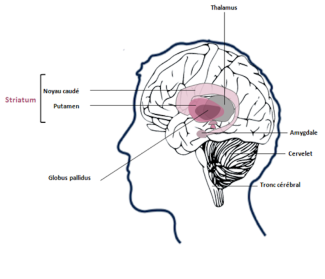
The expansion involves abnormal repetition of a triplet of CAG nucleotides in DNA on chromosome 4. A copy of the protein becomes abnormal and probably toxic to cells when the number of CAG repetitions goes above 35. The huntingtin protein, essential for enabling neurons to survive, is not enough to prevent the disease from occurring. The brain areas affected by atrophy during the disease are the cortex and striatum, which are involved in motor, cognitive and behavioral functions.
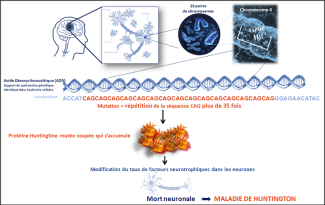
Huntington's disease affects the cortex and the striatum, which are involved in motor, cognitive and behavioral functions.
At Paris Brain Institute
Studying the asymptomatic phase of Huntington’s disease is a key focus of the Basic to Translational Neurogenetics team, co-led by Professor Alexandra Durr. Although patients with Huntington’s disease are born with the expansion, they do not develop symptoms until decades later. What happens in the meantime?
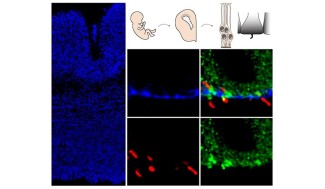
An active collaboration between the clinician-researcher and Dr Sandrine Humbert at GIN in Grenoble has shown that brain anomalies are already present in the fetus. However, the disease is not apparent at birth.
There are signs of cell impairments in the fetus, but no clinical impairment in the child or adolescent, and the clinical signs only appear in adulthood. This suggests that transient compensation for the impairments is occurring. Alexandra Durr’s team aims to understand the mechanisms at play during the disease’s non-symptomatic phase, hoping to activate the compensation processes that delay disease onset.