The pathophysiological mechanisms of ALS that cause motor neuron degeneration are still little understood. The discovery of genetic mutations linked to the disease has identified some of these mechanisms.
The biological mechanisms of Charcot’s disease
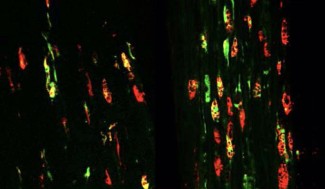
Discovered in 2011, the C9ORF72 gene mutation is present in around 40% of familial forms of ALS, 30% of familial forms of FTD, and 70% of patients affected by both these conditions. In 2006, toxic aggregations of the TDP-43 protein were also identified in these diseases. We now know that they are present in 95% of ALS cases (all forms – familial and sporadic), and 60% of FTD cases. Discovering these aggregates shared by both conditions has provided a molecular basis for this clinical continuum long observed by doctors. All carriers of the C9ORF72 mutated gene also have TDP-43 aggregates, but neither the physiological link between the two nor their interactions have been clearly identified as of yet.

Source: Elisa Teyssou et al., Acta Neuropathologica 2013, 125: 512-522.
Images courtesy of Professor Danielle Seilhean, anatomical pathologist working in Séverine Boillée’s team
Disruption to autophagy?
One of the suspected reasons for the presence of these toxic aggregations is a problem with the processing of waste inside cells. The Ubiquiline 2 (UBQLN2) gene is one of the genes involved in ALS. This is the gene required for processing waste in the neurons. A study by several teams at Paris Brain Institute in partnership with the University of Limoges focused on UBQLN2 gene mutations. Using functional studies, the researchers found that one of the cellular degradation pathways, autophagy, was impaired in patients carrying a Ubiquiline 2 mutation.
Chronic inflammation
For a long time, scientists have known that protein aggregations in the brain, such as amyloid plaques in Alzheimer’s disease and a-synuclein in Parkinson’s disease, are sources of inflammation, but in the case of TDP-43 the picture is unclear.
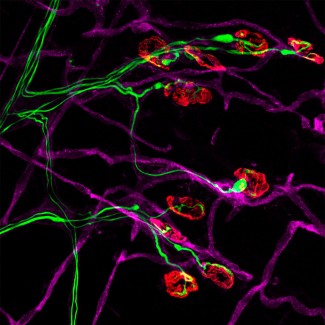
The role of the nervous system’s resident immune cells, microglia, in the disease is now recognized. In ALS, impaired spinal motor neurons have the specific feature of being surrounded by microglial cells in the the motor neuron that exits the spinal column to connect the muscle to the periphery. However, the role of macrophages was, until recently, unconfirmed. In 2020, Séverine Boillée’s team at Paris Brain Institute showed, for the first time, the important role of peripheral macrophages in the progression of ALS. Like microglia, these macrophages generate a chronic inflammatory state for motor neurons. The important feature of macrophages is that they are easier to target from the periphery than microglial cells in the central nervous system.
At Paris Brain Institute
Modelling ALS cellular interactions using patient cells
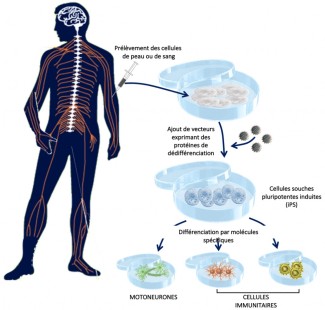
Delphine Bohl’s objective, working within Séverine Boillée’s team, is to model the disease using human induced pluripotent stem cells (iPSCs). This advanced technology can generate any type of cell, including motor neurons and immune cells, from skin cells (via a biopsy) of patients. iPSCs have two key abilities: they are able to multiply infinitely and differentiate into any cell type of the body if exposed to the right signals. These new cell models are a valuable tool that, for the first time, allows us to access patients’ human neurons. The first step is being able to very precisely characterize the motor neurons obtained from the iPSC cells of patients, first in genetic cases, where the mutation that causes ALS is known, and then in sporadic cases, to identify any common mechanisms. The second stage is to bring the motor neuron and immune cells together in a very controlled environment, to model their interactions. Ultimately, these models would also enable the efficacy of therapeutic molecules to be tested.
Protocol for the procurement of induced pluripotent stem cells (iPS) and their derivatives