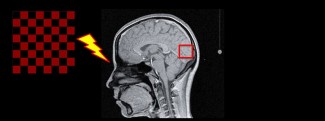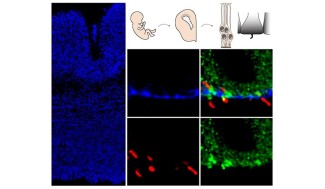
Huntington’s disease, formerly known as Huntington’s chorea, is a rare and hereditary neurological disorder first identified by the physician George Huntington in 1872. It emerges in adulthood as motor, cognitive and psychiatric disorders that worsen over time.
-

18 mille personnes sont touchées en France
Around 10 in every 100,000 people have Huntington’s disease. Onset occurs between the ages of 35 and 50, and prevalence is the same for men and women. In 10% of cases, disease onset occurs before the age of 20.
In France, 18,000 people are affected by this condition, of which 6,000 currently have symptoms and around 12,000 are carriers of the mutated huntingtin gene but are so far asymptomatic.
Biological mechanisms and causes of Huntington’s disease
Huntington’s disease is a genetic and hereditary condition caused by CAG repeat expansion in the gene coding for the huntingtin protein. It is transmitted in an autosomal dominant pattern: one heterozygous copy of the expansion is enough to develop the disease later in life. Descendants of people with this disease have a 50% risk of inheriting this expansion.
Huntington’s disease symptoms and diagnosis
Symptoms of Huntington’s disease often emerge between the ages of 30 and 50, with motor problems, including chorea, and anxiety and depression. These symptoms are mainly noticed by close family and friends, and can be treated with effective symptomatic therapy once symptoms have become established.
Motor symptoms generally include abnormal, abrupt, jerky and involuntary movements of all four limbs, impaired balance and dysarthria. Cognitively, patients often tend to experience a slowing down in their information processing, have planning deficits and, over time, memory and concentration problems.
These disorders are very quickly accompanied by behavioural disorders such as irritability, anxiety and apathy, leading to frontal-subcortical syndrome.
The initial diagnosis is based on the signs exhibited by patients and can be confirmed through genetic analysis, for which the family must be informed in advance. Once the expansion has been identified, affected families can seek genetic counselling to find out whether they are carriers of the mutation, and consequently can undergo prenatal or pre-implantation diagnostics if they are planning a family.
Treatments for Huntington’s disease
There is no cure for Huntington’s disease. However, a range of symptomatic treatments may be offered. These include neuroleptics, dopaminergic inhibitors and anti-depressants, suitable for alleviating motor, psychiatric and sleep problems. Physiotherapy and speech therapy are required and are effective. Psychological counselling is also important for managing the symptoms of the disease and supporting the patient and their friends and relatives. Social support is crucial.
Clinical & translational neurosciences Alexandra DURR’s and Sandrine HUMBERT team focus on neurogenetic diseases, spinocerebellar degenerations – SCD (spastic paraplegias and cerebellar ataxias), frontotemporal lobar degenerations – FTLD and...
Read moreSupport Paris Brain Institute
Did you like this content and did it answer the questions you were asking? Don't hesitate to support Paris Brain Institute.