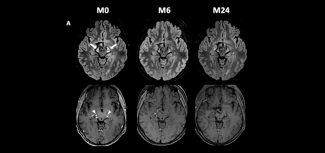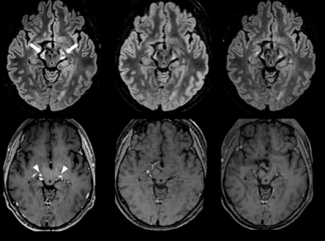
Leukodystrophies are a group of around 100 rare genetic diseases involving damage to the white matter of the central nervous system (brain and spinal cord), with or without changes to the myelin of the peripheral nervous system.
In France, between 150 and 300 children are estimated to be born with leukodystrophy each year.
Biological mechanisms and causes of leukodystrophies
Most leukodystrophies are genetic and more than 200 genes have so far been identified in these conditions. The disease has differing clinical pictures, owing to the mode of transmission of causal mutations in the family and the function of the mutated gene.
Leukodystrophies are caused by genetic anomalies called mutations in the genes that code for proteins that play a key role in the synthesis or maintenance of myelin (a substance that insulates and protects the axons of neurons in the same way as a protective sheath around electrical wires, and which plays an important role in the rapid dissemination of nerve messages from one neuron to another).
Myelin is an ultra-specialised membrane consisting of proteins and lipids that need to be produced and renewed throughout life. Mutations in a gene involved in one of these stages within the glial cells will compromise myelin synthesis and/or maintenance cycles, and will therefore lead to a drop in the quality of myelin or an impairment of myelin functions. Any damage to the myelin causes harm to the neuron, and this can in turn degenerate, leading to a loss of communication between the brain and its periphery, and the onset of motor and sensory symptoms.
Some leukodystrophies are known as hypomyelinating leukodystrophies. This means leukodystrophies in which myelin production is insufficient from birth. Others are demyelinating leukodystrophies, in which the existing myelin is degraded. The latter are generally more severe and progress more rapidly.
Leukodystrophies are generally progressive neurodegenerative diseases and onset can occur in early childhood, adolescence or adulthood.
Leukodystrophy symptoms and diagnosis
Leukodystrophies compromise the function of the myelin sheath. This causes a loss of communication between the neurons and the onset of motor, cognitive, psychiatric and/or sensory symptoms.
The symptoms that patients present differ according to the type of leukodystrophy and the degree of severity of the damage. However, the most common and most frequent clinical signs of motor problems are spasticity (muscle stiffness), ataxia (loss of fine motor coordination of voluntary movements), speech disorders and abnormal movements such as dystonia (involuntary muscle contractions) or tremors. These motor disorders are often coupled with bladder control problems.
Depending on the patient’s age, cognitive symptoms can present as delays in psychomotor development, learning difficulties, difficulties performing duties at work or even dementia. Behavioural or mood disorders are also common. Vision and/or hearing problems are also possible, particularly in children.
Leukodystrophies are diagnosed primarily on the basis of brain imaging (MRI). This analysis makes it possible to highlight anomalies in the myelin and to classify the leukodystrophy as either hypomyelinating or demyelinating. A more accurate diagnosis of the type of leukodystrophy requires biochemical assays for certain leukodystrophies, and genetic testing in all cases. When the diagnosis has been made, family genetic counselling is offered so as to identify any relatives at risk of developing or transmitting the disease to their children and grandchildren.
There are often delays in diagnosing leukodystrophies in adults because the disease can be confused with multiple sclerosis, the most common acquired myelin disease, and the one best known to neurologists. There are specialist rare leukodystrophies centres in France called Centres de Référence de Leucodystrophies Rares, and one focus of their work is to improve diagnosis of leukodystrophies and offer the appropriate care and treatment.
Treatments for leukodystrophies
Dedicated treatments are available for some leukodystrophies, particularly bone marrow transplants for X-linked adrenoleukodystrophy (X-ALD), leukodystrophies with axonal spheroids and pigmented glia (ALSPs) and certain forms of metachromatic leukodystrophy. To be effective, the transplant must be carried out at an early stage of the disease. It is sometimes possible to treat the disease with medicines, for example in cerebrotendinous xanthomatosis. Several gene therapies are also being developed for leukodystrophies.
For all leukodystrophies, symptomatic treatments are possible, involving medicines and/or rehabilitation, to improve symptoms such as spasticity or pain.
In 2019, Prof. Fanny Mochel, a neurologist (Pitié-Salpêtrière Hospital, APHP) and researcher within the MOV’IT team led by Prof. Stéphane Lehericy and Prof. Marie Vidailhet at Paris Brain Institute, performed a haematopoietic stem cell transplant in a patient with rapidly progressing ALSP. By monitoring this patient over 30 months, the symptoms and cerebral lesions associated with the disease were found to have receded.
Link to this publication: DOI: 10.1111/ene.14694
Three types of leukodystrophies researched at Paris Brain Institute
At Paris Brain Instituteresearch focuses on three of the most common types of leukodystrophies:
- X-linked adrenoleukodystrophies (ALD)
- Metachromatic leukodystrophies (MLD)
- Leukodystrophies with axonal spheroids and pigmented glia (ALSP)
Violetta Zujovic’s team is researching the mechanisms of demyelination and remyelination in multiple sclerosis using cells from the immune system. As shown by the success of bone marrow transplants, the immune system plays a key role in certain leukodystrophies. In collaboration with Prof. Fanny Mochel and Dr Caroline Sevin, Violetta Zujovic is now studying the role of the immune system in leukodystrophies in order to identify new therapeutic approaches.
Prof. Mochel’s team is investigating the metabolic regulation of glial cells and how metabolic treatments can slow or even halt certain neurodegenerative processes. The team recently took part in an international therapeutic trial that showed that a molecule targeting energy metabolism and inflammation in X-ALD could reduce the risk of cerebral forms of the disease. Work is currently under way to determine whether this treatment could be an alternative to marrow transplants in X-ALD.
Dr Sevin’s team is using gene therapy to correct the genetic anomaly at presymptomatic stages (before the onset of symptoms) in children with X-ALD, metachromatic leukodystrophy and Krabbe disease.
X-linked adrenoleukodystrophies (ALD)
These diseases affect 1 in 17,000 newborn babies worldwide. It is the most common leukodystrophy. In ALD, demyelination occurs in the central and peripheral nervous system, as well as a dysfunction of the adrenal glands (adrenal insufficiency) – glands located above the kidneys that secrete certain hormones such as adrenaline.
ALD is caused by mutations in the ABCD1 gene located on the X chromosome. The role of the ATP-binding cassette protein (ABCD1) is to break down very long-chain fatty acids (VLCFAs) inside cells. When this protein is no longer functional due to a mutation, VLCFAs accumulate in tissues and become toxic. The level of VLCFAs in the blood is a marker of the disease and aids diagnosis.
Men with XALD pass the disease on to their daughters, but never to their sons. Women with the disease have a 50% risk of transmitting it to their children.
There are three clinical pictures identified for ALD, and they differ in terms of symptoms and patient population.
Form | Age of onset | Symptoms | Treatments |
CALD. ALD of the brain in children, adolescents and adults. The most serious form of ALD. It is characterised by rapid neurological deterioration. The majority of men with ALD are at risk of developing cerebral damage, compared with less than 1% of women. | C-CALD 3-10 years Ado-CALD 11-21 years A-CALD > 18 years | Troubles attentionnels, visuels, auditifs, psychiatriques associés à des troubles de la marche et de la coordination | Treatment of symptoms. In the early stages of the disease, a stem cell transplant may be offered. |
AMN. adrenomyeloneuropathy. Chronic and progressive damage to central and peripheral myelin. Common in women with leukodystrophy and in 100% of men with the disease. | AMN between 20 and 40 for men Between 40 and 60 for women | Problems with walking, balance and bladder and bowel function. Mild paralysis of the lower limbs associated with muscle contractures. | Treatment of symptoms, spasticity, urinary and sphincter disorders and neuropathic pain. |
ISRN. Lower adrenal insufficiency. This condition is characterised by progressive destruction of the adrenal glands. It is found in less than 1% of women with leukodystrophy and in almost 100% of men. | Between 3 and 10 years old All ages | The first visible sign of this condition is a spontaneous tanning of the patient, known as melanoderma. | Treatment by hormone supplementation |
Metachromatic leukodystrophies (MLD)
Metachromatic leukodystrophies are rare autosomal recessive genetic diseases that affect 1 in 45,000 newborns, and that can develop in childhood, adolescence or adulthood.
These leukodystrophies are mainly caused by mutations in the ARSA gene, which codes for a protein responsible for the breakdown of lipids called sulfatides that are essential for the formation and maintenance of the central and peripheral myelin.
Childhood forms of the disease develop before the age of 30 months and cause symptoms such as muscle weakness (hypotonia), language difficulties and motor deficits. As the disease progresses, spasticity, convulsive seizures and visual and hearing problems appear.
Onset for juvenile forms of this disease is between 30 months and 16 years, and can cause behavioural and balance problems.
Adult forms of the disease, which appear after the age of 16, lead to psychiatric and cognitive changes, an increase in mood swings and motor disorders.
Treatments for this disease must be comprehensive and, in some cases, a stem cell transplant may be considered in the early stages.
Leukodystrophies with axonal spheroids and pigmented glia (ALSP)
These leukodystrophies, also called leukoencephalopathies, are caused by a mutation in the CSF1-R gene, which codes for a receptor present on the surface of microglial cells (the immune cells resident in the central nervous system). These are autosomal dominant diseases, meaning that the children of someone with the disease have a 50% risk of also having it. They affect between 10% and 25% of adults with leukodystrophies.
The average age of onset of these leukodystrophies is 40.
The disease affects men and women equally, and presents mainly as rapidly progressing spasticity and/or cognitive and psychiatric disorders. The first signs of the disease generally appear after the age of 30, but it shows incomplete penetrance.
Symptoms include progressive memory loss, changes in personality, motor problems and sometimes a loss of judgement and social withdrawal.
There is currently no specific treatment for ALSPs. Secondary risks associated with this disease need to be monitored, such as recurrent pneumonia or urinary tract infections.
Pelizaeus-Merzbacher disease
Pelizaeus-Merzbacher disease is caused by mutations in the PLP1 gene, which codes for a protein present in oligodendrocytes and which is involved in the synthesis of myelin. This disease mainly affects children, with significant developmental delays that affect motor and cognitive functions. Women carrying mutations in the PLP1 gene may also develop symptoms in adulthood, particularly spasticity and urinary disorders.
Projects at the Paris Brain Institute
Two collaborative projects involving two research teams are under way:
Myelin Plasticity and Regeneration team – Paris Brain Institute. Project leader Violetta Zujovic, INSERM researcher.
Mov’It team – Paris Brain Institute. Project leader: Prof. Fanny Mochel, APHP neurologist.
The Mattrix project
The MATTRIX project studies macrophage activation profiles in X-ALD.
In this disease, acute and chronic inflammation is observed due to the activation of macrophage immune cells.
Macrophages are blood cells (white blood cells) that defend the body against pathogens such as viruses and bacteria. Together with other immune cells such as lymphocytes, they support inflammatory response.
The aim of the MATTRIX project is to understand how mutations in the ABCD1 gene influence the intrinsic capacity of macrophages to become activated and directed towards inflammatory or anti-inflammatory profiles. The findings of this project could lead to more personalised patient care and treatment, with improved predictions for disease progression to help guide first-line treatments.
Inspire project
The INSPIRE project focuses on three types of leukodystrophies (ALSP, MLD and ALD) and aims to:
- Gain a better understanding of the inflammation observed in these diseases, with the aim of identifying new therapeutic targets.
- Contribute to the development of reliable biological criteria to direct patient treatment towards stem cell transplantation as early as possible, since this is currently the only effective therapy for leukodystrophy progression.
- Confirm a new non-invasive treatment for cerebral leukodystrophy (CALD).
- Find new biomarkers for the early detection of neuroinflammation, and gain a better understanding of the biological mechanisms of leukodystrophies by targeting anti-inflammatory response.
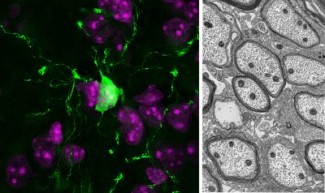
The team aims to understand the neuroinflammatory and metabolic mechanisms involved in multiple sclerosis and leukodystrophies, with a particular focus on the role of cellular metabolism and immune cells in lesion progression and central nervous...
Read moreSupport Paris Brain Institute
Did you like this content and did it answer the questions you were asking? Don't hesitate to support Paris Brain Institute.