Approximately 50% of patients with frontotemporal lobar degeneration (FTLD) have a family history of the disease, suggesting a genetic cause. To date, more than 20 genes carrying mutations have been associated with familial forms of FTD (TC9orf72, GRN/Progranulin, MAPT/TAU, etc.), but some familial forms (approximately 5%) remain unexplained, with no mutations identified. The cause of sporadic forms (without family history) has not yet been determined.
Causes of Frontotemporal Dementia (FTD)
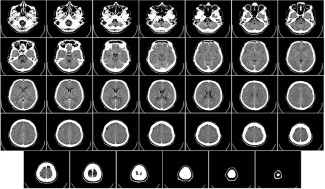
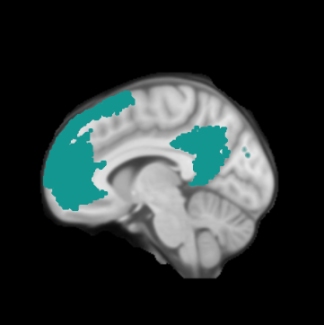
In all cases of FTLD, symptoms are the result of atrophy of the frontal and temporal regions caused by neuronal death specifically in these areas of the brain.
At the cellular level, neuronal death is caused by a build-up of protein in neurons in the frontal and temporal lobes. Most often, it involves an accumulation of the protein TDP-43 (60% of cases), in rare cases the protein TAU (30%), and very occasionally the protein FUS.
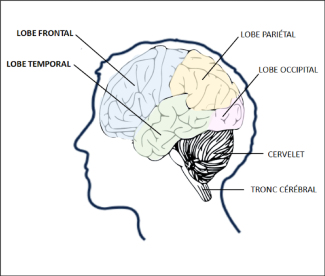
The frontal regions are involved in controlling our behavior, particularly our social behavior and emotions. They are also involved in cognitive functions known as “executive functions”, which include a wide range of abilities such as planning, reasoning, and decision-making. The temporal regions are essential for language and word comprehension.
To find out more about these brain regions, read the work of the FrontLAB: Frontal Functions and Pathology team led by Prof. Richard Levy at Paris Brain Institute:
https://parisbraininstitute.org/news/frontal-lobe-brain-conductor/
At Paris Brain Institute
The search for new genes responsible for FTD is one of the challenges involved in research into these diseases. A study conducted by Dr Isabelle Le Ber (Basic to Translational Neurogenetics team led by Prof. Alexandra Durr and Prof. Giovanni Stevanin) has led to the identification of a new gene, ataxin-2, involved in frontotemporal degeneration. This discovery represents a breakthrough in the diagnosis of these diseases and in genetic counseling for patients’ families.
A study conducted by Isabelle Le Ber’s team shows for the first time the influence of genetics on the age of onset of certain familial forms of frontotemporal dementia. The precise identification of genetic factors should ultimately enable better prediction of the age of onset of the disease in at-risk individuals, which will have a direct impact on genetic counseling and patient care.
Frontotemporal degeneration (FTD) and amyotrophic lateral sclerosis (ALS) are neurodegenerative diseases that can have a common genetic cause, the most common of which is a mutation in the c9orf72 gene. Some preclinical developments targeting this gene offer encouraging therapeutic prospects. In order to test the effectiveness of these potential therapies, it is essential to identify markers that can detect the onset of lesions at an early stage and monitor the progression of the disease.
At Pitié-Salpêtrière Hospital, AP-HP, the PREVDEMALS cohort includes 80 asymptomatic individuals who carry the c9orf72 mutation and are therefore at risk of developing FTD or ALS in the next few years. These individuals were monitored for 36 months (neuropsychological, structural, and microstructural analyses of the brain’s white matter and the cerebral metabolism, biological and clinical examinations) in order to identify prognostic markers for the onset of the disease at the asymptomatic stage.
Studies conducted on this cohort by several teams at the Institute have already shown very early alterations in brain function and structure before the onset of symptoms in people at risk because they carry the mutation.
Data sheet updated with the assistance of Isabelle Le Ber, neurologist, researcher in the FrontLAB team at Paris Brain Institute and coordinator of the reference center for rare and early dementias.