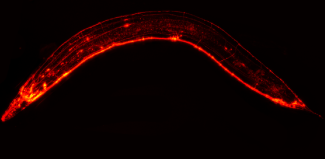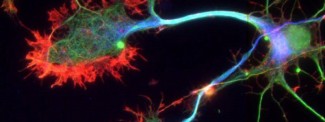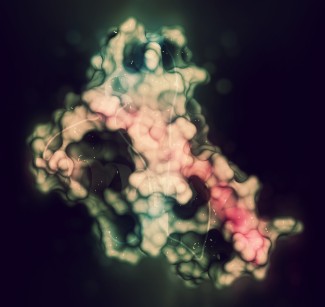
Prion protein or PrP is present in most of our cells. Although highly conserved among species, its role is still poorly understood. In the neurons of sick people, prion proteins adopt an abnormal conformation (misfold), become resistant to degradation, and aggregate together: these are known as “scrapie” prion proteins, or PrPsc. Protein deposits then multiply and accumulate inside and outside neurons, causing them to malfunction and eventually degenerate. The spread of prions depends on their ability to interact with the normal form of the prion protein, change its conformation, and convert it into a pathological form.
At Paris Brain Institute
Most neurodegenerative diseases, from Alzheimer’s disease to Parkinson’s disease and including amyotrophic lateral sclerosis, share common characteristics with prion diseases. The study of prion protein propagation mechanisms provides valuable information about these diseases. The team led by Marie-Claude Potier and Stéphane Haïk, Alzheimer’s Disease and Prion Diseases, is particularly interested in the spread of lesions typical of Alzheimer’s disease, amyloid plaques, which appear to behave similarly to pathological prion proteins. Understanding these “prion-like” mechanisms could benefit both diseases.
To find out more: https://parisbraininstitute.org/news/prion-diseases-model-neurodegenerative-disorders