What if a worm allowed a quantum leap in research on prion diseases such as Creutzfeldt-Jakob disease? A recent study by Nicolas Bizat (University of Paris) and Stéphane Haïk (Inserm) at the Paris Brain Institute, published in the journal Brain, suggests the potential of this model for therapeutic research and identifies five promising molecules for these diseases.
Human prion diseases are fatal neurodegenerative disorders that include sporadic, infectious and genetic forms. Inherited Creutzfeldt-Jakob disease due to the E200K mutation of the prion protein-coding gene is the most common form of genetic prion disease. The phenotype resembles that of sporadic Creutzfeldt-Jakob disease at both the clinical and pathological levels, with a median disease duration of 4 months. To date, there is no available treatment for delaying the occurrence or slowing the progression of human prion diseases. Existing in vivo models do not allow high-throughput approaches that may facilitate the discovery of compounds targeting pathological assemblies of human prion protein or their effects on neuronal survival.
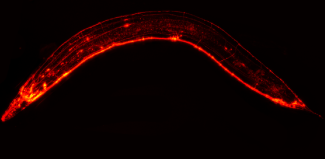
Here, we generated a genetic model in the nematode Caenorhabditis elegans, which is devoid of any homologue of the prion protein, by expressing human prion protein with the E200K mutation in the mechanosensitive neuronal system.
Expression of E200K prion protein induced a specific behavioural pattern and neurodegeneration of green fluorescent protein-expressing mechanosensitive neurons, in addition to the formation of intraneuronal inclusions associated with the accumulation of a protease-resistant form of the prion protein. We demonstrated that this experimental
system is a powerful tool for investigating the efficacy of anti-prion compounds on both prion-induced neurodegeneration and prion protein misfolding, as well as in the context of human prion protein. Within a library of 320 compounds that have been approved for human use and cross the blood–brain barrier, we identified five molecules that were active against the aggregation of the E200K prion protein and the neurodegeneration it induced in transgenic animals.
This model breaks a technological limitation in prion therapeutic research and provides a key tool to study the deleterious effects of misfolded prion protein in a well-described neuronal system.
Sources
https://pubmed.ncbi.nlm.nih.gov/34687213/
Bizat N, Parrales V, Laoues S, Normant S, Levavasseur E, Roussel J, Privat N, Gougerot A, Ravassard P, Beaudry P, Brandel JP, Laplanche JL, Haïk S.Brain. 2021 Oct 22;144(9):2745-2758.