Amyotrophic lateral sclerosis (ALS), or ‘Lou Gerhig’s disease’, is a progressive and fatal neuromuscular condition involving the gradual death of motor neurons, the neurons that control walking, speech, swallowing and breathing. This loss of motor neurons leads to muscular atrophy and progressive paralysis in patients.
-

8 000 personnes touchées en France Environ 450 000 dans le monde
-

55 Ans âge moyen des personnes touchées
-

2.7 nouveaux cas par an pour 100 000 habitantes et habitants
Lou Gerhig’s disease (ALS) is the most common motor neuron disease in adults.
There are two types of motor neuron:
- The central motor neurons, located in a specific region of our brain, the motor cortex, transmit contraction commands to the spinal cord. The peripheral motor neurons, motor neurons located in the spinal cord, transmit motor information to the muscles.
- The degeneration of these two types of motor neuron in ALS leads to a loss of information between the brain and the voluntary muscles, which are therefore no longer recruited, stop contracting and atrophy.
Motor neuron damage can sometimes be associated with a loss of neurons in the characteristic frontal and temporal regions of the brain, leading to cognitive and behavioural problems of varying intensity. In its most severe form, this leads to frontotemporal dementia (FTD): 15% of patients suffering from Lou Gerhig’s disease also have FTD. In FTD, dementia does not include memory problems as it does in Alzheimer’s disease but behavioural changes are typical.
This results in changes to patients’ social behaviour and personality.
Causes of Lou Gerhig’s disease: is it hereditary?
For 1 in 10 patients, the disease originates from hereditary genetic mutations, referred to as familial cases.
Biological mechanisms of Lou Gerhig’s disease
ALS, or Lou Gerhig’s disease, is a disease that affects motor neurons, the nerve cells that control voluntary movements. These motor neurons gradually degenerate, although the precise mechanisms that lead to this neuronal death are not yet known. This neurodegenerative disease progresses within a few years to the point of complete paralysis.
Symptoms, progression and life expectancy with Lou Gerhig’s disease
The symptoms of ALS, or Lou Gerhig’s disease, are complete paralysis of the muscles in the arms, legs, mouth and tongue, as well as respiratory muscles. This leads to an inability to use your arms, or to walk, eat or speak, as well as breathing difficulties, with an onset that is gradual.
This disease begins in adulthood (the average age is 59 in France) and progresses, within 3 to 5 years on average, to complete paralysis of the muscles and eventually to the patient’s death, usually due to respiratory failure. Depending on the location of the motor neurons affected – cortical neurons, brain stem and/or spinal cord – the symptoms may vary between patients, especially at the start of the disease.
Lou Gerhig’s disease treatments and diagnosis
The disease is diagnosed when suspected by a neurologist, who upon neurological examination and investigation finds symptoms that indicate gradual damage to the first- and second-order motor neurons. In all cases, there is gradual loss of muscle strength, sometimes combined with muscle wasting, or stiffness in the limbs. To make the diagnosis, the neurologist uses the results of an examination that records electrical signals between the neurons and the muscles: an electromyogram. These results confirm the motor denervation of the muscles, as a result of motor neuron loss. To confirm this diagnosis, it is vital that the neurologist can, through further tests, rule out all other possible causes of motor neuron damage, such as mechanical compression, inflammatory damage to the motor nerves or any other process that may damage the nerves.
In familial forms of the disease (10% of cases), the diagnosis is made with certainty when the patient carries one of the known mutations responsible for the condition. There are currently more than 30 known genes, and at least one third of familial cases still have no genes identified (the proportion varies between world regions).
At present, there is no cure for Lou Gerhig’s disease. However, combining neuroprotective treatment with multidisciplinary care will ensure that treatment covers all aspects of the disease and can slow the progression of symptoms.
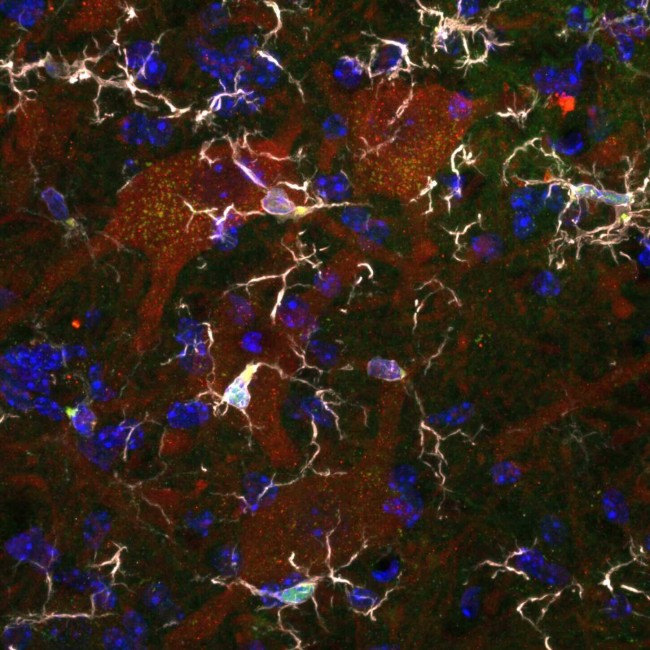
The team led by Séverine BOILLEE is interested in the mechanisms that induce motoneuron degeneration in ALS and that may result from a deleterious interaction between these neurons and microglial and macrophage cells (immune system cells).
Read more
The team "Molecular Physiopathology of Parkinson’s Disease" is interested in the complexity of Parkinson’s disease and conducts multidisciplinary research projects combining clinical evaluation, neuronal biology and genetics.
Read more
The goal of the Frontlab team is to understand better the role and organization of the prefrontal cortex in the control, activation, and inhibition of goal-directed behaviours.
Read moreSupport Paris Brain Institute
Did you like this content and did it answer the questions you were asking? Don't hesitate to support Paris Brain Institute.