Hereditary spastic paraplegias are a heterogeneous group of pathologies and share clinical similarities with other neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS).
For the first time, Khalid H. El Hachimi and his collaborators in Alexis Brice's team, showed neuropathological similarities (at the level of the nervous system lesions) between type 11 spastic paraplegia and ALS. This discovery will enable clinicians to make an accurate and early diagnosis facing an " atypical " ALS, via research of a mutation in SPG11 gene. On the other hand, the understanding of the mechanisms involved in these pathologies paves the way for the development of new targeted therapeutics.
Hereditary spastic paraplegias are a heterogeneous group of diseases on clinical, genetic, and neuropathological levels. The most common inherited autosomal recessive form, is caused by a mutation in SPG11 gene. Clinical signs gradually settle down and are characterised by very debilitating walking disorders due to lower limbs stiffness or spasticity. This clinical presentation can be complicated by various symptoms shared with other neurological diseases such as amyotrophic lateral sclerosis, neuropathies or cerebellar ataxias. From a physiopathological point of view, this disease is characterised by degeneration of the cortico-spinal beam (neurons are located in the motor cortex and their axons are projected onto the spinal cord) controlling voluntary motor skills.
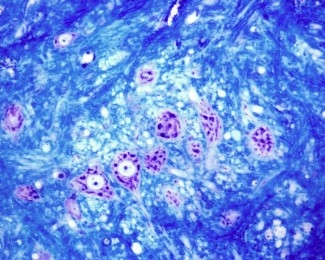
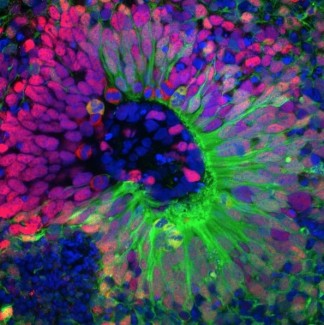
For the first time, Khalid H. El Hachimi and his collaborators in Alexis Brice's team, showed neuropathological similarities (at the level of the nervous system lesions) between type 11 spastic paraplegia and ALS. Through the post-mortem study on patients' brains, researchers have observed lesions (ubiquitin and P62) at the level of the spinal cord and medulla oblongata similar to those found in ALS. These specific inclusions in motor neurons are also associated to an accumulation of lipidic products. The protein encoded by SPG11 gene, called spatacsin is involved in vesicular and axonal transport, calcium homeostasis regulation and in lysosome recycling. The loss of function of spatacsin would lead to a lysosome recycling defect and an intra-neuronal accumulation of non-degraded products, which could be associated to neuronal death.
These studies illustrate neuropathological similarities between spastic paraplegias and ALS, and highlight the pathophysiological continuum of motor neuron degeneration. These results also contribute to a better understanding of lysosomal dysregulations and could open new paths for the development of therapeutic solutions.
Reference : Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions.