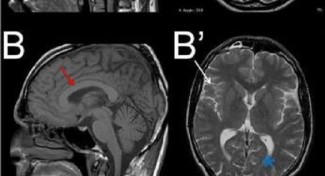
Hereditary Spastic Paraplegia (HSP) are a genetically heterogeneous group of invalidating neurological disorders characterized by a progressive difficulty walking due to stiff legs (spastic) and additional neurological signs like epilepsy, ataxia, mental retardation, primary and secondary motor neuron involvement, etc, that complicate the clinical picture which then overlap with other CNS pathologies (amyotrophic lateral sclerosis [ALS], cerebellar ataxias, peripheral neuropathies,…) .
Hereditary Spastic Paraplegia (HSP) are a genetically heterogeneous group of invalidating neurological disorders characterized by a progressive difficulty walking due to stiff legs (spastic) and additional neurological signs like epilepsy, ataxia, mental retardation, primary and secondary motor neuron involvement, etc, that complicate the clinical picture which then overlap with other CNS pathologies (amyotrophic lateral sclerosis [ALS], cerebellar ataxias, peripheral neuropathies,…) .
The team of Giovanni Stevanin, INSERM and EPHE researcher in Institut du Cerveau - ICM (Director: Professor Alexis Brice), works on genetic and physiopathological mechanisms implicated in these diseases.
To date, mutations in >40 genes, named SPG for spastic gait gene followed by a number in order of their discovery, can account for these diseases. In agreement with early functional and neuropathological investigations, the first causative genes were implicated in intracellular trafficking. The novelty in recent years is the increasing implication of lipid metabolism in these disorders. Indeed, the INSERM / EPHE team at Institut du Cerveau - ICM has identified 4 new causative genes in less than one year, all encoding enzymes of the lipid metabolism.
The last identified gene by Giovanni Stevanin and collaborators, is the B4GALNT1 / SPG26 gene which encodes for -1,4-N-acetyl-galactosaminyl transferase 1 (B4GALNT1). Using the next generation sequencing approach, they identified 7 mutations in 7 large families with HSP. , all leading to a loss of function of the B4GALNT1 enzyme. This fits well with the neurological impairment observed in the b4galnt1 knock-out mouse in which the accumulation of simple gangliosides and the absence of complex gangliosides are observed.
The increasing involvement of lipid metabolism in HSPs, and especially SPG26, will benefit to the patients since the fact that ubiquitous enzymes are affected lead to the possibility to detect abnormal lipid profiles in peripheral blood samples in order to facilitate clinical diagnosis and pave the way for future therapeutic trials since it is possible to act at various levels of the involved metabolic pathways. In addition, the fact that complex gangliosides are known to be critical elements of membranes and particularly in rafts, connects the two major functional pathways involved in HSP: lipid metabolism and intracellular trafficking. Finally, patients with lipid metabolism defects are often presenting with complex phenotypes that overlap with other CNS diseases such as leucodystrophy, therefore highlighting clinical overlaps between these diseases and also with non-CNS disorders since some patients are infertile.